

A genetic heart rhythm disorder linked to mutations in the KCNH2 gene
What is KCNH2-related Long QT Syndrome?
KCNH2-related Long QT Syndrome (LQT2) is a genetic heart rhythm disorder caused by mutations in the KCNH2 gene, which affects how the heart beats. It belongs to a group of conditions known as Long QT Syndromes (LQTS), where the electrical activity in the heart takes longer than normal to reset between beats, resulting in a prolonged QT interval on an electrocardiogram (ECG).
This delayed repolarization can increase the risk of dangerous arrhythmias, fainting (syncope), seizures, or even sudden cardiac death, especially when triggered by stress, emotions, or loud noises.

What causes KCNH2-related Long QT Syndrome?
The condition is caused by mutations in the KCNH2 gene, which provides instructions for making a potassium ion channel protein involved in resetting the heart’s electrical rhythm. When this channel doesn’t function correctly, it slows down the electrical recovery process after each heartbeat, leading to a prolonged QT interval.
It is inherited in an autosomal dominant pattern, meaning a child can inherit the condition from just one affected parent. However, it can also occur as a spontaneous (de novo) mutation.
Who is at risk?
- Individuals with a family history of Long QT Syndrome or sudden cardiac death
- Children and adults with a known KCNH2 gene mutation
- Females tend to have a higher risk of symptoms and complications than males
- Those exposed to QT-prolonging medications, electrolyte imbalances, or intense emotional or physical stress
What are the symptoms of KCNH2-related Long QT Syndrome?
Symptoms can vary in severity, and some individuals may be asymptomatic. Common symptoms include:
- Fainting (syncope), especially after emotional stress, physical exertion, or sudden loud noises
- Seizures, often misdiagnosed as epilepsy
- Palpitations or feeling of skipped heartbeats
- Sudden cardiac arrest, particularly in undiagnosed or untreated individuals
- Some may show no symptoms, but still have an abnormal ECG and genetic findings
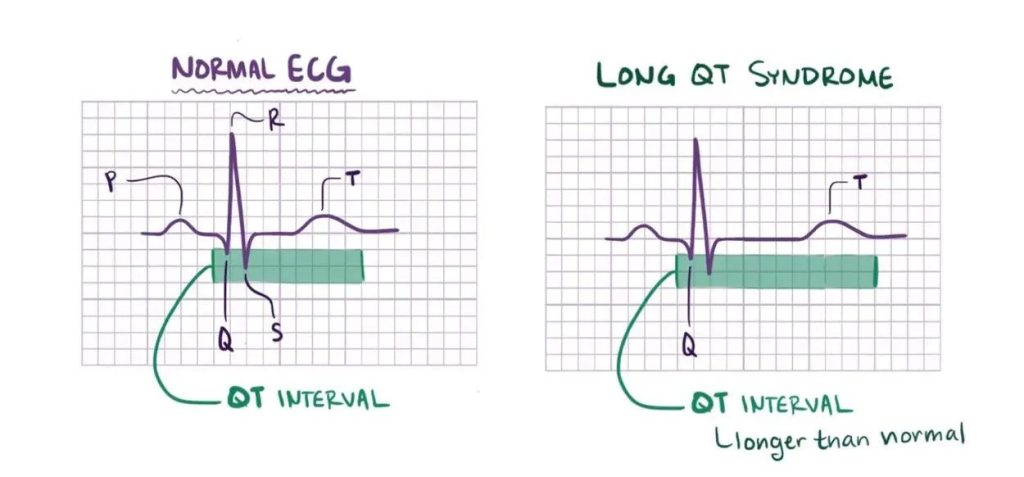
How is KCNH2-related Long QT Syndrome diagnosed?
Diagnosis involves a combination of clinical evaluation, heart tests, and genetic testing:
- Electrocardiogram (ECG) – shows prolonged QT interval
- Holter monitor or event recorder – to capture irregular heart rhythms over time
- Genetic testing – to confirm mutations in the KCNH2 gene
- Exercise or auditory stress testing – may help reveal rhythm abnormalities
- Family screening – is crucial when a diagnosis is made
How is KCNH2-related Long QT Syndrome treated?
Treatment is aimed at preventing abnormal heart rhythms and reducing the risk of sudden death:
- Beta-blocker medications – first-line treatment to control heart rhythm and prevent arrhythmias
- Implantable cardioverter-defibrillator (ICD) – may be recommended for high-risk individuals or those who have survived cardiac arrest
- Avoiding QT-prolonging drugs, such as certain antibiotics, antidepressants, and antihistamines
- Lifestyle adjustments – like avoiding intense sports, emotional extremes, or sudden noise exposure
- Potassium and magnesium supplementation – if electrolyte imbalances are detected
What is the prognosis for KCNH2-related Long QT Syndrome?
- With early diagnosis and appropriate treatment, most individuals can live long and active lives
- Beta-blockers and ICDs greatly reduce the risk of life-threatening arrhythmias
- Ongoing monitoring and family screening are important for long-term management
- Some cases may remain asymptomatic but still require precautionary treatment
Can KCNH2-related Long QT Syndrome be prevented?
While the genetic mutation cannot be prevented, early detection and intervention are key to preventing complications:
- Genetic counseling is recommended for affected families
- Prenatal or preimplantation genetic testing may be considered in families with a known mutation
- Education about avoiding triggers and QT-prolonging medications can reduce risks significantly